DNA becomes damaged upon exposure of cells to high-energy radiation and mutagenic chemicals that are prevalent in our environment. These agents, along with replicative stress and endogenous oxygen radicals and reactive aldehydes, pose a constant threat to the maintenance of a stable genome. Of the myriad DNA lesions, the DNA double-strand break (DSB) is among the most harmful because of its potential to cause deletions and other gross chromosome rearrangements. Accordingly, failure to properly resolve DSBs can lead to cellular transformation, cancer, and other pathologies, such as intellectual impairment and neurological disorders. Dysregulation of DSB repair pathways is a major cause of innate and acquired resistance to cancer therapy as well.
Elucidation of DSB repair mechanisms will exert a major impact on health sciences, and will also provide insights to guide protection against DNA damage, explain drug resistance in cancer therapy, and identify new targets for the development of novel therapeutics tailored to the DSB repair status in cancer patients.
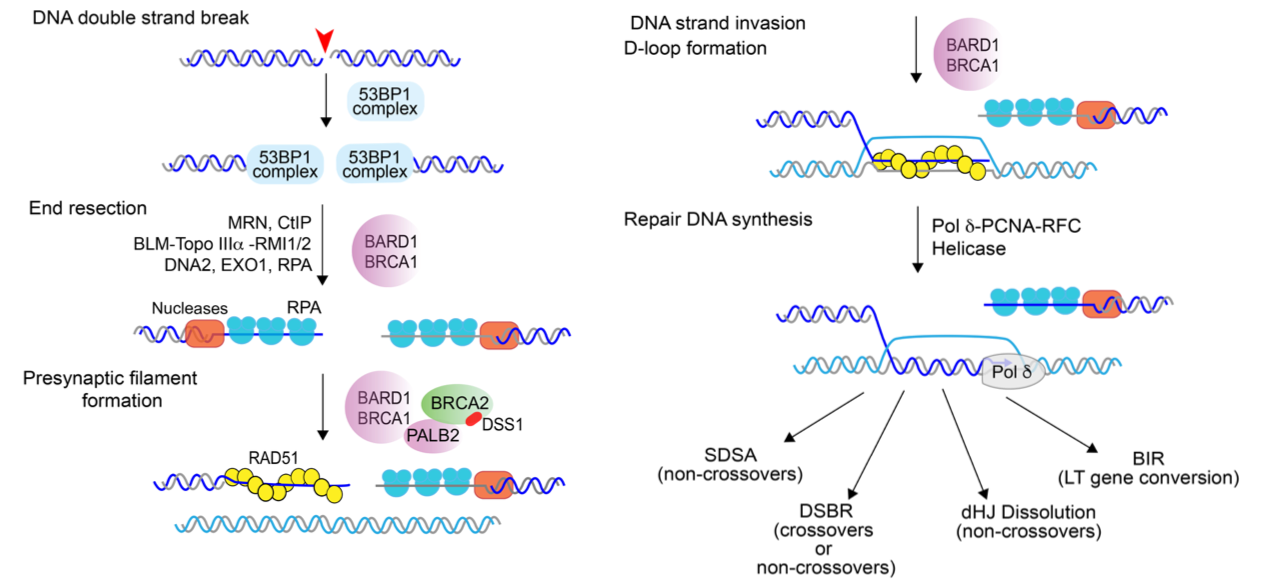
Our laboratory investigates the mechanistic underpinnings of DSB repair mechanisms, particularly in the homology-directed DNA repair (HDR) pathway which plays a critical role in the error-free elimination of DSBs and in replication fork repair (Figure below). Our research harnesses a variety of biochemical, biophysical, and cell biological analytical tools to interrogate HDR factors at the molecular and cellular level, and to draw a comprehensive picture of the HDR machinery.
Currently, we are actively working on the following projects;
1) Regulation of DSB repair pathway choice:
Germline or acquired mutations in BRCA1 (Breast Cancer Gene 1) are clinically associated with the development and progression of breast and ovarian cancers. Cells deficient in BRCA1 or harboring mutant versions of the protein are defective in HR as a result of impaired DNA end resection and an inability to load the recombinase RAD51 onto ssDNA derived from resection. In contrast, loss of anti-resection factors including 53BP1 and its interacting partner RIF1 leads to a partial restoration of DNA end resection and the ability to conduct HR in BRCA1-deficient cells.However, this apparently favorable outcome of HR restoration paradoxically renders cells resistant to PARP inhibitors, the newest class of anti-cancer drugs that are efficacious against BRCA- and other HR-deficient breast cancers. How exactly does 53BP1 or RIF1 loss in BRCA1-deficient cells promote resistance to PARP inhibitors remains undefined.
Recently, the multi-subunit Shieldin complex, comprising of SHLD1, SHLD2, SHLD3, and REV7/MAD2L2, has been found to be recruited by 53BP1-RIF1 to DSBs, where it ′shields′ the DNA ends from unregulated resection. As such, Shieldin facilitates the engagement of NHEJ as the default repair pathway by preventing the channeling of DSBs into HR. Studies in human cells show that loss of Shieldin impairs NHEJ and leads to a hyper-resection phenotype, as indicated by an increase in levels of phosphorylated RPA as well as RPA nuclear foci, a reliable surrogate marker for DNA end resection, whereas its overexpression impairs HR. Moreover, mutations in genes encoding the Shieldin subunits promote resistance to PARP inhibitors in BRCA1-deficient cells, implicating its likely importance in the management of BRCA1-mutant tumors. Therefore, delineating the mechanism of action of Shieldin in DSB repair pathway choice will yield insights into how this pathway choice is regulated in humans and exert a significant impact on the fields of breast cancer biology and DNA repair.
2) Processing of RNA/DNA hybrids and R-Loop resolution by Senataxin-BRCA1 axis:
Senataxin (SETX), an RNA-DNA helicase is mutated in a juvenile type of amyotrophic lateral sclerosis (ALS4) and ataxia with oculomotor apraxia 2 (AOA2). Such neuropathies are believed to stem from defects in RNA processing and transcription termination functions of SETX. Spontaneous DSBs generated during transcription pose a major threat to genomic stability in post-mitotic neurons. This is evident by the association of multiple neuronal disorders with genetic defect in DNA repair genes such as NBS1 and ATM.
Recent reports of accumulation of RNA at DSBs, and an RNA-templated HR process in post-mitotic neurons warrant further investigation of the relevance of RNA in DSB repair. SETX accumulates at DSBs in a transcription-dependent manner, and associates with BRCA1 and other DDR factors. SETX depletion leads to an increase in 53BP1 foci and chromosomal translocations via NHEJ. We are currently invested in determining the association of SETX and other proteins involved in RNA metabolism with the HR repair pathway.
3) Regulation of DNA end resection
4) Mechanism of tumor suppressor proteins BRCA1, BRCA2, PALB2, RAD51 and paralogs, and their associated proteins in HDR and DNA replication fork protection:
5) Mechanism of Fanconi anemia proteins in HR